Le syndrome de Rett typique (MIM #312 750) est une encéphalopathie neurodéveloppementale très particulière, touchant essentiellement les filles, caractérisée, dans sa forme typique, par une décélération globale du développement psychomoteur, puis d’une perte des acquisitions cognitives et motrices, survenant après une période de développement normal. Il constitue la principale cause de retard mental chez la fille, avec une prévalence variable de 1/10 000 à 1/22 000 naissance féminine dans les différentes régions du monde.
Le diagnostic clinique repose sur des critères cliniques qui ont été modifiés avec les années avec l’accroissement des connaissances sur les symptômes de la maladie (Neul et al., 2010; Naidu and Johnston, 2011 ; Hagberg et al., 2002). Malgré tout, les éléments clés du diagnostic sont toujours présents qui permettent de poser le diagnostic de syndrome de Rett typique ou classique. Parallèlement au syndrome de Rett typique un certain nombre de patientes présentaient certains symptômes clés du Rett, mais pas l’ensemble des signes de la maladie. Ces formes ont été dénommées « Rett atypiques ou variantes ». Selon les symptômes associés, on a ainsi pu définir trois formes facilement reconnaissables : la forme avec préservation du langage, la forme avec épilepsie précoce et la forme congénitale (Hagberg and Skjeldal, 1994).
En 1999, des mutations dans le gène codant pour Methyl-CpG-binding protein 2 (MECP2) ont été identifiées dans d’exceptionnelles formes familiales du syndrome de Rett et validées dans des formes sporadiques (Amir et al.,1999). Aujourd’hui on sait que les mutations et délétions de MECP2 sont retrouvées dans 95 à 97 % des cas des formes typiques. Par contraste, dans les formes atypiques ou variantes, on ne retrouve une mutation de MECP2 que dans 50 à 70 % des cas. Plus récemment des mutations dans d’autres loci que celui de MECP2 ont été identifiées dans des formes atypiques de Rett. Parmi celles-ci, des mutations dans CDKL5 ont été identifiées dans les formes atypiques avec épilepsie précoce (Bahi-Buisson et al., 2008). De la même manière, des mutations dans FOXG1 ont été retrouvées chez des enfants porteurs de formes congénitales (Ariani et al., 2008).
Le syndrome de Rett est un « concept clinique », qui repose sur l’association de caractéristiques cliniques coexistant chez une même patiente, une séquence d’évolution particulière, et l’exclusion de certains diagnostics différentiels {Hagberg, 1994 #247 ; 1988 #248}. Ces caractéristiques ont été définies par Hagberg dans les années 80 pour les formes typiques ou classiques (3/4 de cas) et dans les années 90 pour les formes atypiques ou variantes (1/4 des cas) et récemment révisées en 2010 (Tableau 1). L’un des critères essentiels du syndrome de Rett classique est la perte documentée, habituellement entre 1 et 2 ans, d’acquis psychomoteurs, tels que l’utilisation des mains, le babillage et les capacités de communication. Cette période de régression qui peut durer quelques semaines à quelques mois, est suivie d’une période de « réveil », où on assiste à une récupération relative des capacités de communication visuelle. Ces éléments permettent de différencier fondamentalement le syndrome de Rett des pathologies dégénératives progressives du système nerveux central.
On distingue des critères nécessaires, accessoires ou d’appui au diagnostic et des critères d’exclusion.
Tableau 1 - Les critères révisés des syndromes de Rett
Critères de diagnostic du syndrome de Rett 2010
Envisager le diagnostic quand il existe une décélération postnatale de la croissance du périmètre crânien
Critères nécessaires pour le diagnostic de Rett typique ou classique
Une période de régression suivie par une phase de récupération ou stabilisation*
Tous les critères majeurs et éliminer tous les critères d’exclusion
Les critères accessoires ne sont pas requis, bien que souvent présents dans le syndrome de Rett typique
Critères nécessaires pour le diagnostic de Rett atypique ou variant
Une période de régression suivie par une phase de récupération ou stabilisation*
Au minimum 2 des 4 critères majeurs
Au minimum 5 des 11 critères accessoires
Critères majeurs
Perte partielle ou complète de l’utilisation volontaire des mains
Perte partielle ou complète du langage oral **
Troubles de la marche: Instable (dyspraxique) ou absence de marche
Stéréotypies manuelles caractéristiques : pression/torsion, applaudissement, frottement/lavage des mains
Critères d’exclusion pour le diagnostic de Rett typique ou classique
Lésions cérébrales d’origine périnatale ou postnatale, troubles métaboliques ou maladie neurologique progressive ***
Développement psychomoteur anormal pendant les 6 premiers mois de la vie ****
Critères accessoires *****
Dysfonctionnement respiratoire avec des épisodes d’apnée pendant la veille, d’hyperventilation intermittente, d’épisodes de blocage de la respiration, d’expulsion forcée d’air ou de salive
Bruxisme pendant la veille
Troubles du sommeil
Troubles du tonus
Troubles vasomoteurs
Scoliose/cyphose
Retard de croissance
Mains et pieds petits, hypotrophiques
Rires ou accès de cris ou de pleurs inappropriés
Diminution de la sensibilité à la douleur
Communication par le regard intense - “eye pointing“
* Étant donné que les mutations dans MECP2 sont parfois identifiées chez des enfants avant la régression, on parle d’un diagnostic de Rett « possible », chez les enfants de moins de 3 ans qui n’ont pas présenté de régression, mais qui présentent d’autres signes évocateurs de Rett. Ces enfants doivent être évalués tous les 6 mois. Si une régression se manifeste, le diagnostic de Rett est « défi nitif ». Toutefois, si un enfant ne présente pas de régression avant 5 ans, le diagnostic de Rett doit être discuté.
** La perte du langage acquis porte sur le niveau acquis avant la régression, babillage, bissyllabisme ou mots, ou phrases.
*** Il est impératif d’exclure toute pathologie neurologique infectieuse, traumatique ou métabolique ou dégénérative (par un examen clinique, et une imagerie scanner ou IRM).
**** On parle de retard majeur des acquisitions psychomotrices (absence de tenue de tête, absence de sourire réponse, troubles de déglutition). Une hypotonie modérée et un décalage des acquisitions dans le syndrome de Rett ne constituent pas un critère d’exclusion.
***** Il s’agit de signes cliniques que l’enfant a présentés un jour et pas nécessairement au moment de la consultation. Une large proportion des critères accessoires dépendent de l’âge, et peuvent être très prévalents en fonction de l’âge. De ce fait, le diagnostic de Rett atypique est parfois plus difficile à établir chez les plus petits. Chez les plus petites (< 5 ans) qui ont présenté une période de régression et ≥ 2 critères majeurs, et qui n’ont pas 5/11 critères accessoires, on parle de « Rett atypique probable ». Ces enfants doivent être réévalués régulièrement.
Ces différents signes permettant de suspecter le diagnostic s’inscrivent dans un profil évolutif particulier caractéristique de l’affection. (Tableau 2) Ainsi, quatre périodes se succèdent après une « phase silencieuse » où le développement est normal. Une première phase de stagnation précoce qui débute entre 6 et 18 mois. Habituellement, cette phase dure quelques semaines à quelques mois et se caractérise par un retard des acquisitions psychomotrices, mais sans réelle régression. La seconde phase de « régression neurologique rapide », entre 1 et 4 ans. Cette phase peut être très rapide et durer quelques jours à quelques semaines, mais peut aussi se prolonger pendant plusieurs mois. La régression touche les acquisitions motrices et mentales, et s’associe à une perte très caractéristique d’utilisation des mains coïncidant avec l’apparition de stéréotypies manuelles. À ce stade, le principal diagnostic différentiel est l’autisme infantile. La troisième phase dite « pseudo-stationnaire », ou de stabilisation apparente qui débute entre 2 et 10 ans, concerne essentiellement les filles ayant acquis la marche. En effet, durant cette période, on assiste à une régression de certaines aptitudes motrices, comme la marche, alors qu’il existe une récupération relative des capacités de communication (contact visuel, notamment) et une diminution des traits autistiques. Toutefois, la majorité des filles Rett garde une « pseudomarche » (qui ressemble à une déambulation de type pyramidale) jusqu’à l’âge adulte. La quatrième phase de « détérioration motrice tardive » débute à partir de l’âge de 10 ans. Pour les filles n’ayant jamais acquis la marche, elles passent directement du stade 2 à 4. Cette phase se caractérise, pour les enfants ayant marché, par une perte de la marche, et l’apparition de nouveaux symptômes spécifiques et invalidants, tels que les troubles du tonus (dystonie, hypotonie) et des déformations squelettiques (principalement une scoliose). En revanche, malgré cette régression motrice, le contact visuel et la socialisation sont conservés pendant toute la période adulte.
Tableau 2 - Les quatre stades cliniques du syndrome de Rett
Stade 1 : Stagnation d’apparition précoce
Début : entre 6 et 18 mois
Arrêt ou stagnation du développement psychomoteur
Diminution de l’intérêt pour les jeux
Hypotonie
Ralentissement de la croissance céphalique
Durée : plusieurs mois
Stade 2 : Régression rapide
Début : entre 1 et 4 ans
Régression des capacités de communication avec manifestations autistiques, détérioration du comportement, automutilation, perte de l’usage des mains, du babillage / langage
Stéréotypies manuelles
Convulsions (15 %)
Durée : quelques semaines à quelques mois
Stade 3 : Stabilisation apparente pseudo-stationnaire - phase de « réveil »
Début : entre 2 et 10 ans
Récupération d’un contact visuel et régression des manifestations autistiques
Stéréotypies manuelles caractéristiques, apraxie/dyspraxie manuelle prédominante
Régression motrice lente avec pseudo-marche, spasticité, ataxie, apraxie
Durée : quelques mois à quelques années
Stade 4 : Évolution tardive
Après 10 ans
Détérioration de la motricité avec perte de la marche
Scoliose, atrophie musculaire
Syndrome pyramidal et extrapyramidal marqué
Retard de croissance
Troubles trophiques
Amélioration du contact visuel
Absence de langage
Épilepsie moins sévère
Durée : plusieurs années
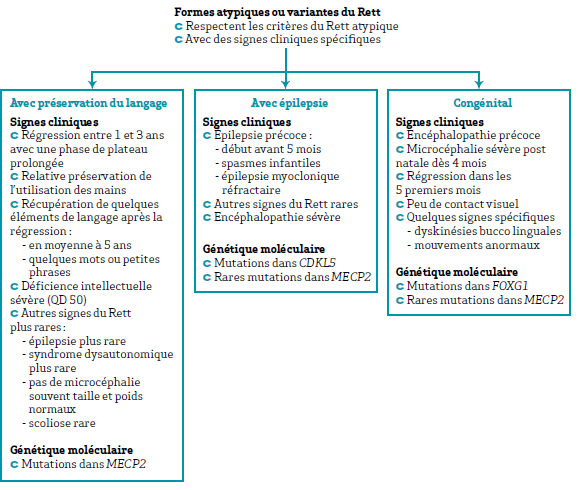
Une grande variété des formes atypiques ou variantes du Rett ont été identifiées sur la base de signes cliniques et évolutifs communs. Ces formes répondent à des critères cliniques tout comme la forme classique ou typique.
Les plus fréquentes et reconnaissables sont : la forme avec préservation du langage, la forme congénitale la plus grave qui se présente comme une encéphalopathie précoce, sans phase de développement normal et une forme avec une épilepsie sévère et précoce (< 6 mois). Alors que la forme avec préservation du langage est le plus souvent liée à des mutations dans MECP2, les causes génétiques des formes congénitales et avec épilepsie précoce sont distinctes. Ainsi des mutations dans CDKL5 rendent compte des formes avec épilepsie précoce, alors que des mutations dans FOXG1 ont été identifiées dans des formes congénitales.
La figure ci-dessous illustre les signes cliniques ainsi que les gènes majeurs impliqués dans ces formes atypiques ou variantes du syndrome de Rett.
Conclusion
Plus de dix ans après la découverte de l’association des mutations dans MECP2 et le syndrome de Rett typique, ces « nouveaux critères » permettent une clarification des diagnostics cliniques et donnent un cadre pour les formes atypiques ou variantes.
Avec l’avancée rapide des connaissances sur le syndrome de Rett, l’augmentation croissante des diagnostics précoces par la mise en évidence de mutations dans MECP2 chez des enfants qui ne présentaient pas l’ensemble des critères de la maladie (selon les critères de 2002), la révision des critères du diagnostic des formes typiques et variantes était devenue impérative. Ainsi, avec la définition de situations telles que les syndromes de Rett « probables » et la nécessité de réévaluer les critères régulièrement jusqu’à ce que l’enfant réponde bien à la définition du Rett typique, les cliniciens et les familles ont des repères plus précis dans cette maladie développementale qui a malgré tout une évolution caractéristique. En outre, ces critères ont l’avantage majeur de définir des populations homogènes pour l’inclusion dans des études cliniques et des essais thérapeutiques.